
Medicines Registration - NMRC

REQUIREMENTS FOR MEDICINES REGISTRATION
Applications for registration of human medicines involve the submission of a completed application form together with a dossier of supporting documents in the Common Technical Document (CTD) format.
Applications for registration of veterinary medicines involve the submission of a completed application form together with a dossier of supporting documents in the format known as VMRF or the Common Technical Document (CTD) format.
All applications for registration should be accompanied by the dossier, including the following:
- Electronic submission of documentation (CD or DVD) should be submitted for all CTD modules (1-5) or all VMRF parts in text-selectable PDF format and Microsoft Word (required for templates/summaries, e.g. QOS–PD, BTIF, BW).
- One sample of the smallest pack size for human products and one sample for veterinary products.
- Screening fees. All fees are stipulated in the statutory fees schedule published in 2021, Government Gazette 7608-Gov N178, and they can be downloaded from the E-Library.
REGISTRATION PROCESS
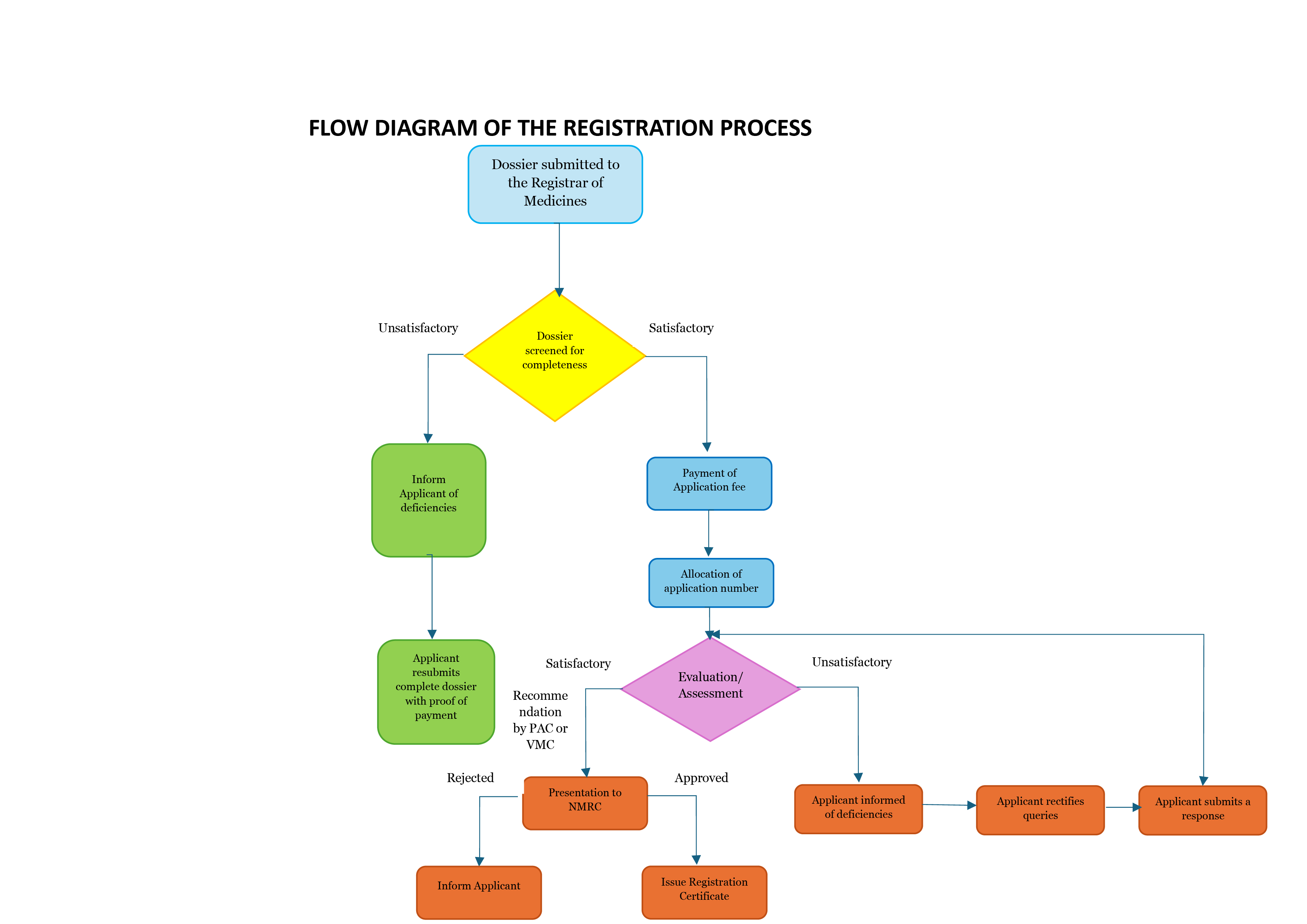
- Medicine application dossiers are received and screened for completeness using the appropriate screening checklist and a response of the outcome of the screening will be communicated to the applicant.
- Applications that pass screening will require payment of application fee to be allocated an application number and progress to the evaluation stage. In the event that an application is incomplete and fails the screening stage, the applicant can re-submit a complete application at a later date. This re-submission will incur another screening fee.
- A detailed review of the dossier is done and a report is generated. Deficiencies, missing information etc. is communicated to the applicant and a deadline to respond to queries outlined (i.e. 90 days from the date of the letter for applicant to respond).
- Completed evaluation reports and recommendations are forwarded to the Pharmaceutical and Analytical Committee of the NMRC or in case of Veterinary medicines to the Veterinary Medicines Committee of the NMRC.
- Products approved by NMRC based on the recommendations of the Pharmaceutical and Analytical Committee or the Veterinary Medicines Committee are allocated a registration number and gazetted. The product is then entered into the Medicines Register and notification of registration communicated to the applicants.
- Recommendations from these committees to Council may define whether a product is to be registered or not.
ALTERNATIVE REGISTRATION PATHWAYS
1. Expedited “Fast-track” Registration Procedure
The Council may fast track review of applications for products as contemplated under regulations 45. More resources are outlaid towards processing applications submitted through the expedited review pathway thus higher fees are prescribed in the current fee schedule.
2. Stringent Regulatory Authorities (SRA) pathway
The Council may fast track review of applications for products as registered in stringent regulatory authorities that the NMRC aligns with. More resources are outlaid towards processing applications submitted through the Stringent Regulatory Authority (SRA) pathway thus higher fees are prescribed in the current fee schedule. The list of stringent regulatory authorities and organizations that NMRC aligns with can be located in the E-library.
3. WHO Collaborative Registration Procedure
This is a WHO Collaborative Registration Procedure for prequalified products. This collaborative procedure serves to facilitate and accelerate registration of products which have already been assessed and prequalified by WHO Prequalification Team-Medicines (WHO/PQTm). All applicants with WHO prequalified products are encouraged to use this route.
Applicants wishing to use this route should submit an expression of interest and notify WHO PQ of their intention to use this procedure for registration of a particular product as outlined on the WHO PQ website: https://extranet.who.int/prequal/
4. ZAZIBONA Collaborative pathway
ZAZIBONA is a SADC collaborative registration initiative among the nine actively participating national medicines regulatory authorities (NMRAs), namely Botswana, Democratic Republic of Congo (DRC), Malawi, Mozambique, Namibia, South Africa, Tanzania, Zambia and Zimbabwe. The remaining seven SADC Member States participates in the collaborative process as non-active members, because they do not contribute assessment report but use the reports from the process in their country regulatory processes. The objective is to facilitate access to good quality medicines through work-sharing in assessment of applications for registration and inspection of manufacturing and testing facilities.
When a product has been approved by Zazibona, the individual countries can rely on this decision at country level to subsequently register/not register a product in the country based on local regulatory requirements.
Applicants interested in having their products assessed under the ZAZIBONA initiative should submit their applications to administrator@zazibona.com or visit the ZAZIBONA website at https://zazibona.com/dossier-assessments/our-process-flow/pilot-procedure/ for more information.
POST-REGISTRATION AMENDMENTS/VARIATIONS
A Marketing Authorization Holder (MAH) is responsible for the registered FPP throughout its life-cycle, irrespective of the regular reviews by the NMRC. It is acknowledged that technical and scientific progress may necessitate changes to the registered product over time. Any changes to a registered FPP (variations), whether administrative or substantial, are subject to approval by NMRC.
Guidance for the implementation of the different types of variations subject to approval are set out in the Guideline for post-registration amendments/variations to approved registration dossiers (version 01).
Alternate approaches to the principles and practices described in the guideline may be acceptable provided they are supported by adequate justification. These approaches should be discussed in advance with NMRC. In addition, it must be noted that NMRC reserves the right to request information or material, or define conditions not specifically described in the NMRC variation guideline, in order to allow for adequate assessment of safety, efficacy or quality of the pharmaceutical product.
MEDICINES REGISTRATION
- 15 Ruhr Street, Northern Industrial Area
- +264-61-203 2400
- +264-61-203 2401
- +264-61-203 2402
- +264-61-203 2465
- Medicines.Nmrc@mhss.gov.na
FREQUENTLY ASKED QUESTIONS (FAQs)
1. How long does the registration process typically take?
- There are currently no specific timelines for the registration of medicines, owing to the current backlog of applications for registration, post-registration amendments and other conflicting obligations. The NMRC is actively engaging with stakeholders and partners to clear the backlog of applications to make provision for the review and assessment of medicine dossiers.
2. What are the requirements for companies outside Namibia intending to register products with the NMRC?
- It is important to note that you are required to appoint a local representative for your product(s) in Namibia. The local representative will facilitate communication with the NMRC regarding any particulars related to products from your company. As regulators, it is unfortunately out of our jurisdiction to appoint or recommend a local representative on your behalf.
- Should you require assistance in appointing a local representative, you may contact our professional pharmacy bodies/organizations such as the Pharmaceutical Society of Namibia (PSN) (https://www.psn.com.na/index.php/about-us) and Pharmacists Care Association (PCAN) (https://www.linkedin.com/in/pharmacists-care-association-of-namibia-pcan-245386264?lipi=urn%3Ali%3Apage%3Ad_flagship3_profile_view_base_contact_details%3BMy%2FBumXJSOO7QJBTtp8RqA%3D%3D ) about your intention to register medicines in Namibia and your need for a local representative. You may contact them via email at: secretary@psn.com
3. Does one need an import license to import sample products for registration?
- No. One does not need an import license to import samples for medicines registration in Namibia. As per the NMRC regulations, only one sample per strength of the product, in the smallest pack size is required for registration purposes.
4. Kindly confirm if Type IN and AN Variations that do not require a new registration certificate, is still exempted from payment, or is N$ 1500 payment now required?
- IN and AN variations that do not require changes to registration certificates are exempted from payment.
5. When an applicant has already submitted 2 strengths of a product and they have both passed screening and the application fees have been paid and would like to add another line extension. Do they need to submit a full CTD application- or a line extension amendment.
- The applicant will be required to submit a full CTD application and pay screening fee. According to the variation guideline under “15.0 Examples of changes that make a new application necessary”, change in the dose and/or strength of one or more APIs results in the submission of a new application.
6. Is a Bio-Equivalence (B.E) study mandatory?
- Yes, a BE study is mandatory in regulatory processes to ensure that a generic drug is equivalent to the innovator/comparator drug in terms of dosage form, safety, strength, route of administration, quality and intended use.
7. If a Bio-Equivalence (B.E) Study can not be provided, can a Bio-waiver be used instead?
- A biowaiver in regulatory settings can be considered when a drug product meets specific criteria, allowing you to avoid in vivo bioavailability and bioequivalence studies. This is generally accepted for immediate-release solid oral dosage forms (like tablets or capsules) that rapidly dissolve and have high solubility.
- The decision to pursue a biowaiver rather than a bioequivalence (BE) study is based on factors such as:
- Drug solubility and dissolution: The drug should be highly soluble and rapidly dissolve in the gastrointestinal tract. (Should be a BCS Class I or III)
- Dosage form: Immediate-release solid oral formulations are most likely to qualify.
- Excipient compatibility: The excipients used should not impact dissolution or absorption
- Drug products having a narrow therapeutic index are excluded from consideration for a BCS-based biowaiver.
8. How many times a year does the Council sit?
- The Namibia Medicines Regulatory Council sits four times a year; once every quarter.
9. Does NMRC regulate medical devices?
- Currently NMRC does not currently regulate medical devices, except under the following circumstances:
- If the device has an active pharmaceutical ingredient, then the device in question, follows the conventional medicine registration route.
- For Covid Antigen test kits, that requires NMRC's approval.
The NMRC is currently working on guidelines for regulating medical devices.